FAQs
What is the genetic code?
Genetic code refers to the instructions contained in a gene that tell a cell how to make a specific protein. Each gene’s code uses the
four nucleotide bases of DNA: adenine (A), cytosine (C), guanine (G) and thymine (T) — in various ways to spell out three-letter “codons”
that specify which amino acid is needed at each position within a protein.
(source: https://www.genome.gov/genetics-glossary/Genetic-Code)
Do I have to register to look at the available data?
No, anyone has access to the available data.
Do I have to register to add new data?
Yes, to add new amino acids or genetic codes, a user account is mandatory. Go to LOGIN to register.
How can users add a new genetic code?
After you are logged in, you can add a new genetic codes by providing the following information:
- full name,
- short name,
- codon table,
- optional references,
- and optional comments.
How can users add a new (non-canonical) amino acid?
After you are logged in, you can add a new amino acid by providing the following information:
- full name,
- short name,
- canonical SMILES code,
- clogD7 value (for methyl ester of N-acetylamino acid),
- and optional comments.
What is clogD7 and how can I calculate its value for an amino acid?
For our calculations, we choose the clogD7 scale from the ChemAxon website (https://disco.chemaxon.com/calculators/demo/plugins/logd/) which is one of the most widely used public databases for calculating the partitioning values. clogD7 indicates the partitioning of a substrate between octanol and water buffered at pH 7 (Figure 1A), and the value is calculated from an empiric dataset. We thus can assign any amino acid structure with a clogD7 value and use this for our scale.
The clogD7 value should be calculated not for a free amino acid but for its derivative as shown below:
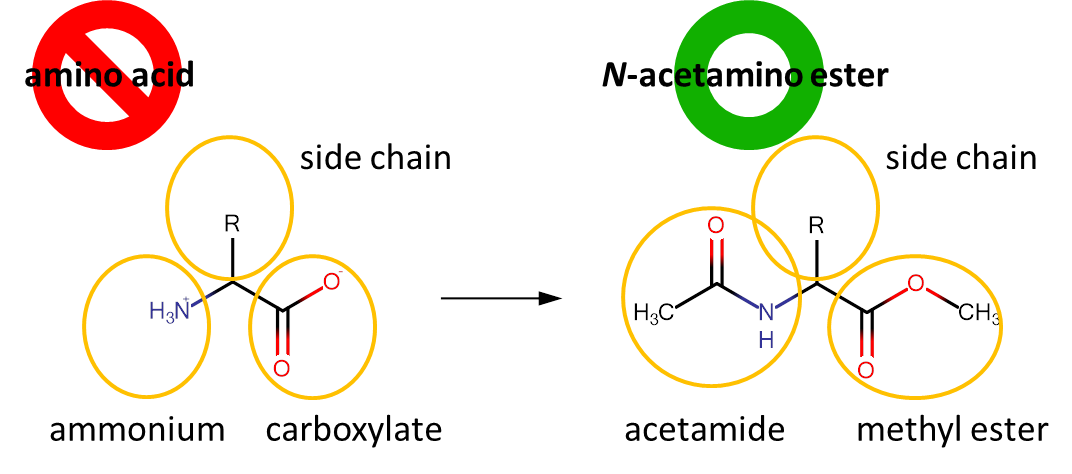
A free amino acid is a zwitter-ion, which is extremely hydrophilic and partitions to the water phase almost entirely. By drawing a derivative, methyl ester of N-acetylamino acid, the user nullifies the hydrophilic contribution of the zwitter-ion, thus the effect of the side-chain group exchange can be seen very well. As a simple rule, the residues that produce a clogD7 value above zero are can be considered hydrophobic, while residues with the clogD7 below zero are hydrophilic. Even slightest changes in the chemical structure will be reflected in the outcome value. For example, hydrophobic phenylalanine residue can be made more hydrophobic or even made hydrophilic depending from the additional groups introduced in the structure:
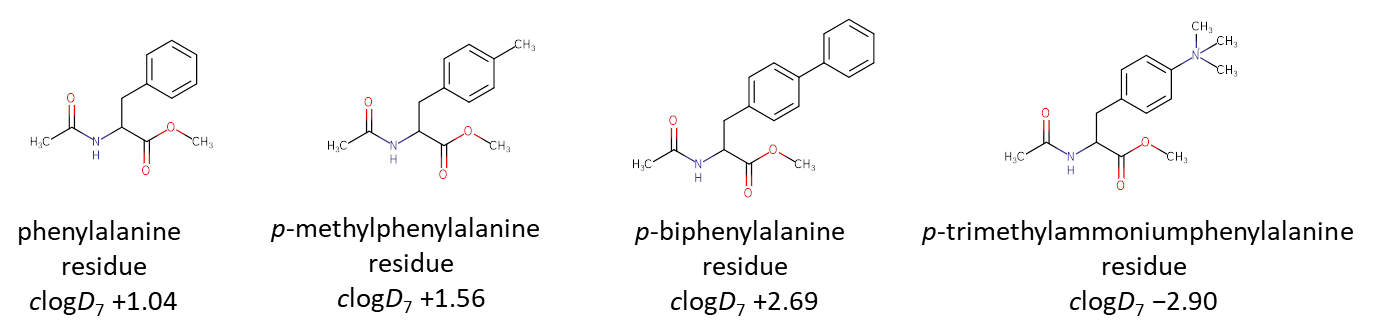
The great thing about the clogD7 value, it can be calculated for amino acid residues, canonical or non-canonical, just by drawing a structure and letting the program calculate the number. The experimental scale have also been developed recently, and experimental logP/logD7 values have been meassured for a number of amino acids and can be found in (Kubyshkin 2021).
What is omitted by the polarity scale using clogD7?
There are few problems with the polarity scale. One of them is the impact of stereochemistry which barely reflects in the experimental logP/logD and typically does not reflect in the calculated values.
Another problem is taking into account the backbone changes. Most of the canonical amino acids share same backbone features with only the side chains varying. Thus, we can follow polarity changes by introducing or removing parts of the backbone structure and reading out the concomitant changes in the polarity value. However, when chemical structure of the backbone gets altered the polarity effects become difficult to enumerate. For example, an exchange of alanine with proline alters the backbone folding. As the result of different exposure of the backbone to the surrounding medium, polarity changes can be quite dramatic despite seemingly close clogD7 values assigned to proline (−0.34) and alanine (−0.61). It is yet unclear how to enumerate the backbone changes and transfer those on the polarity scale. However, the fragment based clogD7 polarity comparison is generally not fully adequate when the backbone is being affected. For example, introduction of secondary amino acids of β-amino acids won’t be well reflected by the proposed scale.
What are SMILES codes and how to generate them?
SMILES (simplified molecular-input line-entry system) is a widely used system for writing chemical structures. A SMILES notation allows concise writing a chemical structure using a simple set of symbols in just one line. A SMILES codes are extremely useful especially in generating and operating chemical databases and calculations. The user is required to provide SMILES code for an amino acid structure in the free (non-rerivatized) form. The codes can be generated by any modern chemical drawing software or in internet resources.
What is the ∆code value?
The method we choose to calculate the distance, or dissimilarity, between two codes is expressed according to
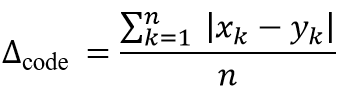
where xk is the (clogD7) value for the kth amino acid in code x and yk the (clogD7) value for the kth amino acid in code y, while n the total number of compared codons is set to 64 (see figure 2).
There are two ways to calculate ∆code:
- The first compares all codon pairs and ignores those pairs where one or both codons encode a stop signal (which means n=61 in the formula used above, when both codes have 3 stop codons at the same position). The advantage is, that we only compare amino acids, for which the clogD7 value is available. The disadvantage is that we run into problems when we include codes where one has a stop signal and the other one an amino acid assigned at the same codon, as these pairs are not included in the calculation.
- The second way to calculate ∆code is to compare all 64 codons (which means n=64 in the formula used above). For those pairs where one is a stop signal and the other is an amino acid a placeholder value is used. The advantage is that we can now compare codes irrespective of where its stop codons are located. The disadvantage is that we introduce an uncertainty into the ∆code value as the placeholder value is not (yet) an exact measure for the difference between a sense and a stop codon. In the calculations on this website we use the second option. The placeholder value used, and which is considered as the lower bound for “actual” value, is the largest difference between any two amino acid as used in (Schmidt and Kubyshkin 2021) which was between phospho-serine (clogD7=-4.89) and mirystyl-glycine (clogD7= 5.24) and which is 10.13.
Here, we decided to use option 2.
How can I navigate in the 3D visualization?
You can use the mouse to navigate the 3D visualization. Click and drag to rotate, zoom in and out or click on a specific code (node) to learn more about it.
Can I download all ∆code values between all available codes?
Yes, you can! Go to the download section and get it.
I want to help improve or promote the website, how I can I do so?
Please get in touch with us by sending us an Email to
How can I learn more about ∆code, clogD7 and the genetic firewall?
The idea of dissimilarity between natural and engineered organisms is described in (Marliere 2009), the concept of xenobiology and the genetic firewall is described first in (Schmidt 2010). The methodology behind ∆code was introduced with canonical amino acids only (Schmidt 2019), it was then further developed and recalculated with clogD7 values to incorporate non-canonical amino acids (Schmidt and Kubyshkin 2021). A comparison between computationally generated clogD7 and experimentally verified logD7 values showed the validity of clogD7 (Kubyshkin 2021).
References:
- Kubyshkin, V. (2021). "Experimental lipophilicity scale for coded and noncoded amino acid residues." Org Biomol Chem 19(32): 7031-7040.
- Marliere, P. (2009). "The farther, the safer: a manifesto for securely navigating synthetic species away from the old living world." Syst Synth Biol 3(1-4): 77-84.
- Schmidt, M. (2010). "Xenobiology: a new form of life as the ultimate biosafety tool." BioEssays 32(4): 322-331.
- Schmidt, M. (2019). "A metric space for semantic containment: Towards the implementation of genetic firewalls." Biosystems 185: 104015.
- Schmidt, M. and V. Kubyshkin (2021). "How to quantify a genetic firewall? A polarity-based metric for genetic code engineering." Chembiochem 22(7): 1268-1284.